
Механизмы развития сердечной недостаточности при хронической болезни почек
Aннотация
Сердечная недостаточность (СН) широко распространена и связана с высокой смертностью при хронических заболеваниях почек (ХБП). Тем не менее патофизиология сердечной дисфункции при ХБП, особенно при латентном течении болезни почек, недостаточно изучена. Инвалидизация и высокая смертность больных с кардиоренальным синдромом (КРС) диктует необходимость изучения механизмов развития сердечной недостаточности при хронической болезни почек. В этом обзоре поиска литературы использовалась база данных PubMed и Google Scholar для выявления механизмов развития сердечной недостаточности при хронической болезни почек. Изучение научных данных за последние 5 лет показало, что кардиоренальный синдром, имеющий сложную и многофакторную патофизиологию, представляет собой клиническую проблему. Диагностические, прогностические и терапевтические меры при кардиоренальном синдроме ограничены. Современные фармакологические методы лечения эффективны, но недостаточны для удовлетворительного воздействия или смягчения прогрессирования кардиоренального синдрома, поэтому открытие новых лекарств и новых терапевтических стратегий КРС является высокоприоритетной задачей. Лечение пациентов с кардиоренальным синдромом должно быть комплексным и непрерывным, направленным на устранение физических и психосоциальных симптомов.
Ключевые слова: хроническая сердечная недостаточность, кардиоренальный синдром, патогенез, коморбидная патология
К сожалению, текст статьи доступен только на Английском
1. Introduction
The cross-interaction of the heart-kidney-vascular network consists of complex biological processes, in which these organs interact synergistically to maintain basic physiological functions [Raina et al., 2020]. Cardiorenal syndrome (also known as renocardial syndrome) is an umbrella term that includes heart or kidney dysfunction that adversely affects another organ and ultimately leads to failure of both [Chinnappa et al., 2017; Melnyk, 2017; Uduman, 2018]. The harmful effect of one organ on another can be direct or indirect and includes a complex feedback system that includes regulatory hormones, inflammatory molecules, and responses to oxidative stress [Coresh, 2017; Savira et al., 2020]. Although the concept of cardiorenal communication is widespread, the underlying molecular mechanism is not clearly defined. However, accumulating evidence indicates that the nervous system, humoral and cellular immune system are involved [Saito, 2020]. The concept of serious adverse renal cardiovascular events (MARCE) is useful to heal researchers better understand the prevalence of cardiorenal syndrome and its use as an endpoint in current research [Ishigami et al., 2020].
2. Pathophysiology of Cardiorenal syndrome
2.1. Inter-organ changes in the pathophysiology of cardiorenal syndrome
Hyperactivation of the renin-angiotensin-aldosterone system RAAS, sympathetic nervous system SNS and disruption of the natriuretic peptide system (NPS) initiate phenotypic and other molecular changes in the heart and kidneys vasculature. These changes cause organ dysfunction, which leads to systemic consequences, which, in turn, affect other organs due to bi-directionality [Kamyshnikova et al., 2017; Vakulenko, 2019].
2.2. Interaction of neurohormonal pathways and pathways of the sympathetic nervous system
The renin-angiotensin system (RAAS) has long been considered the main regulator of blood pressure, but has recently been recognized as a mechanism for modulating inflammation. During inflammation and oxidative stress in kidney disease, hyperactivation of the classical branch of the renin-angiotensin (Ang) system (RAAS) and the sympathetic nervous system (SNS) occurs [Costanzo, 2020].
Angiotensin II is a major component of the renin-angiotensin system (RAS). The angiotensin-converting enzyme (ACE) produces it from angiotensin I. Remarkably, there is a positive feedback loop in the renin-angiotensin system through which angiotensin II can increase ACE levels. Angiotensin II also leads to increased secretion of aldosterone, which causes sodium retention and potassium excretion.
Two main angiotensin II receptors that are expressed in many cells. When angiotensin II binds to the first of these receptors, called the angiotensin 1 receptor (AT1), expressed on fibroblast, cardiac myocytes, smooth muscle cells, kidney cells, adrenal cells, macrophages, microglia, hepatocytes, endothelial cells, etc., vasoconstriction occurs and activation of the transcription factor of the nuclear factor NF –Kb (the transcription factor NF-Kb is a universal transcription factor that controls the expression of genes for the immune response, apoptosis and the cell cycle) and the release of pro-inflammatory cytokines [Takahama, Kitakaze, 2017].
When angiotensin II binds type 1-angiotensin receptors AT1 on podocytes, it induces apoptosis. When it activates these receptors (AT1) on endothelial cells, it leads to increased expression of tissue factor (TF), an essential component of the blood coagulation cascade, as well as vascular permeability and extravasation of neutrophils into tissue.
The inclusion of a second angiotensin receptor, AT2, also quite ubiquitously expressed by angiotensin II, leads to vasodilation and suppression of inflammation. In both acute and chronic inflammatory conditions of the kidneys, the level of the AT1 angiotensin-1 receptor increases and the AT2 type-2 receptor decreases.
A second ACE angiotensin-converting enzyme, ACE2, exists as a membrane-bound protease in many cell types, splitting angiotensin II to form a small peptide ang1-7, which binds to the protein of the Mas receptor to induce an anti-inflammatory and anti-apoptotic program also causing vasodilation. Interestingly, a high concentration of aldosterone decreases Mas expression, also disrupting this link in the renin-angiotensin system of the RAS.
Initially, increased activity of the second angiotensin-converting enzyme (ACE) 2 supposedly helps reduce inflammation, prevent immune damage, and promote renal tissue repair.
However, in a chronic inflammatory process, the compensatory capabilities of the renin-angiotensin-aldosterone system of the RAAS are disrupted and all those systemic negative manifestations that we observe in the form of arterial hypertension, the development of CHF, etc., develop [Takahama, Kitakaze, 2017].
Decreased cardiac output leads to renal hypoperfusion. This, in turn, causes sodium retention to maintain plasma volume by activating the renin-angiotensin-aldosterone system (RAAS). Elevated sodium levels cause constriction of the glomerular arterioles, decreasing the glomerular filtration rate (GFR). In turn, to maintain GFR, vasoconstriction of efferent arterioles is required to increase glomerular filtration pressure [Kumar et al., 2019; Rangaswami et al., 2019]. However, vasoconstriction further reduces renal perfusion and, if prolonged, causes kidney damage due to hypoxia.
Aldosterone production also causes maladaptive sodium reabsorption in the distal renal tubules, resulting in volume overload and expansion of extracellular fluid.
By itself, Angiotensin II (Ang II), as a key neurohormone of the renin-angiotensin-aldosterone system of the RAAS, causes water and sodium retention by increasing the expression of sodium transporters in the proximal renal tubules. Ang II also promotes the production of aldosterone, which acts on mineralocorticoid receptors in the distal renal tubules and collecting ducts, promoting sodium retention. In addition, both Ang II and aldosterone stimulate cardiac fibrosis by stimulating fibroblast growth and collagen synthesis. In addition, cardiac myocytes undergo hypertrophy to compensate for hemodynamic disturbances and increased levels of neurohormones [Kumar et al., 2019].
Hyperactivity of sympathetic nervous system (SNS) is the main compensatory mechanism for maintaining cardiac output and inotropic support. As chronic kidney disease progresses, the SNS is over-activated in response to renal ischemia, an increase in angiotensinogen 2 Ang II, and a decrease in nitric oxide (NO), resulting in hypertension, left ventricular hypertrophy (LVH), and left ventricular dilatation. At the molecular level, hypertrophy, apoptosis and necrosis of cardiac myocytes associated with inflammation and activation of free radical oxidation (ROS) are observed as a direct effect of catecholamines. Persistent activation of myocardial β 1 –adrenergic receptors leads to impaired transmission of the receptor signal. The increased SNS activity also causes vasoconstriction and decreases renal blood flow, which triggers the release of renin. Moreover, the transport of sodium in the proximal tubules is increased due to the enhancement of the apical Na +/ H + antiporter through the simulation of β-adrenergic receptors.
The antidiuretic hormone (or vasopressin), is released through the activation of baroreceptors or indirectly through the sympathetic nervous system (SNS), is another vital hormone that leads to fluid retention. Antidiuretic hormones activates vasopressin 1A (V 1A) receptors on vascular smooth muscle cells to cause vasoconstriction and activates vasopressin 2 (V 2) receptors in the distal tubules to facilitate diffusion of water from the tubular lumen into the interstitium [Savira et al., 2020].
Increased salt/water retention due to activation of the renin-angiotensin-aldosterone system RAAS/SNS also leads to increased intra-abdominal pressure and venous congestion [Agrawal et al., 2019]. Venous congestion in the form of increased central venous pressure reduces the pressure gradient in the capillary network, reducing perfusion. In the context of cardiorenal syndrome, this leads to congestion, glomerular dysfunction and impaired natriuresis. Increased intra-abdominal pressure also decreases renal function by decreasing GFR and plasma flow [Kumar et al., 2019].
Together with the renin-angiotensin-aldosterone system RAAS and SNS, the natriuretic peptide system is also recognized as an important neurohormonal system that maintains cardiorenal homeostasis. In particular, the natriuretic peptide system consists of four endogenous hormones: atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), C-type natriuretic peptide (CNP), and urodilatin [Han et al., 2020]. Atrial natriuretic peptide ANP and brain natriuretic peptide BNP are mainly produced in the heart with urodilatin synthesized in the kidney, and these natriuretic peptides (NP) activate the transcriptional receptor γ-coactivator-1α (PGC-1α) or receptor-peroxisome alpha (pGC-A), while type C natriuretic peptide (CNP) is mainly synthesized in the endothelium and kidneys and activates the beta peroxisome receptor (pGC-B) [Seliger, 2020]. Upon activation of the pGC peroxisome receptor, a second messenger, cyclic guanosine monophosphate (cGMP), is produced, which produces widespread beneficial cardiovascular and renal effects, including vasodilation, natriuresis, diuresis, and inhibition of fibrosis, and cardiomyocyte hypertrophy.
The natriuretic peptide system, the renin-angiotensin-aldosterone system RAAS and SNS are highly integrated systems between which there are significant cross-links, and the natriuretic peptide system is counter-regulating for both the renin-angiotensin-aldosterone system RAAS and SNS, which usually work together. Indeed, the natriuretic peptide system has the ability to inhibit RAAS and SNS and vice versa (Fig. 1) [Savira et al., 2020]. Thus, the pathophysiology of cardiorenal syndrome is classically viewed as a result of an imbalance between the RAAS and/SNS and the natriuretic peptide system.
2.3. Inflammation
Along with hemodynamic and neuroendocrine mechanisms, the main links involved in the development of kidney damage are oxidative stress, activation of the inflammation system, and apoptosis.
In inflammatory kidney diseases, tissue damage attracts inflammatory cells, i.e. monocytes, and increases the production of cytokines, especially IL-1, IL-6 and TNF- α, to the damaged area [Harrison et al., 2020]. Physiologically pro-inflammatory molecules are required to stabilize the site of injury and the formation of scar tissue. However, prolonged activation can lead to pathological fibrosis and endothelial dysfunction, aggravating renal vasoconstriction and leading to glomerulosclerosis and arterial hypertension. Increased expression of tumor necrosis factor (TNF- α) and IL-6 in the kidney is associated with the activation of the signaling pathways of the nuclear factor κB (NF-κB) transcription factor, the main regulator of cellular inflammation.

Fig. 1. Interaction of neurohormonal pathways and pathways of the sympathetic nervous system [Savira et al., 2020]
Рис. 1. Взаимодействие нейрогормональных путей и путей симпатической нервной системы [Savira et al., 2020]
2.4. Oxidative stress
The inflammatory process in the kidneys stimulates oxidative stress (OS), that is, excess production of oxidants compared to stabilizing antioxidants (nitric oxide) [Sagoo, Gnudi, 2018].
The kidneys are a very energetic organ. This makes them more vulnerable to damage by the OS. In turn, OS is associated with the progression of kidney disease. In addition, complications of chronic kidney disease (CKD), such as inflammation and cardiovascular disease (CVD), the leading cause of death in CKD patients, are also associated with increased OS levels.
The so-called “oxidative” link between CKD and its complications is achieved through several mechanisms, such as uncoupling of endothelial nitric oxide synthase (eNOS) caused by uremic toxin and increased activity of nicotine amide adenine dinucleotide phosphate oxidase [NADPH oxidase (NOX)], also loss of antioxidants due to dietary restrictions, use of diuretics, loss of energy with protein and/or decreased intestinal absorption [Virzì et al., 2015; Daenen et al., 2019].
Extensive experimental and clinical data accumulated over the past 3 decades have demonstrated that increased oxidative stress in CKD occupies a key position as a central link in complex and interrelated pathways involved in the pathogenesis of CKD [Duni et al., 2017; Barrows et al., 2019]. Excessive production of reactive oxygen species (ROS) under conditions of activation of several enzyme systems such as nicotine amide adenine dinucleotide phosphate (NADPH) oxidase, lipoxygenase, xanthene oxidase, unbound nitric oxide synthase (NOS), and mitochondrial respiratory chain along with impaired mechanisms antioxidant defenses (for example, superoxide dismutase (SOD), catalase, selenium-containing glutathione peroxidase and paraoxonase (PON)) underlie the imbalance between enhanced prooxidant and insufficient antioxidant capacity that occurs in CKD, further leading to oxidation of macromolecules, tissue damage and dysfunction. [Pavlakou et al., 2017; Zhang et al., 2017]. Thus, the excessive production of reactive oxygen species by ROS was directly associated with the initiation and progression of CKD, which subsequently leads to arterial hypertension and diabetes mellitus. In addition, the triad of oxidative stress, chronic micro-inflammation (which implies a state of persistent subclinical inflammation) and endothelial dysfunction as a hallmark of CKD supports and perpetuates the vicious circle in which chronic kidney damage occurs, leading to systemic complications of CKD, in particular, cardiovascular dysfunction.
Increased oxidative stress in CKD is traditionally associated with loss of renal function and the use of renal replacement therapy (hemodialysis or peritoneal dialysis) in patients with end-stage CKD [Liakopoulos et al., 2019; Roumeliotis et al., 2019; Chrysohoou et al., 2020]. Thus, both increased production of reactive oxygen species (ROS) and decreased clearance of prooxidant substances under conditions of renal dysfunction, together with impaired antioxidant arsenal, are responsible for the pro-oxidant environment that characterizes CKD.
However, it should be noted that oxidative stress is already present even in the early stages of CKD with increased production of nicotine-amide-adenine-dinucleotide-phosphate-oxide-dependent superoxide NADPH-oxidase-dependent superoxide by inflammatory cells in the circulation. Moreover, research models of ischemic reperfusion injury suggest that oxidative stress may also act as a link between acute kidney injury (AKI) and the progression of CKD. Biomarkers that characterize the transition of acute renal injury from acute renal failure to CKD, such as urine thioredoxin, a protein that regulates redox potential, or serpin A3/alpha-1-antichymotripsin and angiotensin in urine, are the subject of extensive ongoing research [Zhang et al., 2017].
Oxidative stress markers such as plasma F2-isoprostanes, 8-oxo-7,8-dihydro-2'-deoxyguanosine, malonyl dialdehyde (MAD), advanced oxidation protein products (AOPP) and carbamylated proteins, and asymmetric dimethylarginine (ADMA) show that particles of oxidized lipoproteins accumulates in CKD as renal dysfunction progresses [Schei et al., 2017; Efremova et al., 2020].
Albuminuria is a well-known marker of kidney damage. It occurs in the early stages of many forms of CKD and is the main factor in the progression of the disease due to the induction of both mesangial and tubular toxicity, as well as the activation of internal renal and systemic inflammatory pathways [Ronco et al., 2018; Coresh et al., 2019]. A dysfunctional glomerular filtration barrier with damage to podocytes underlies the development of proteinuria and subsequent glomerulosclerosis. The mechanisms underlying podocyte damage are complex and include hemodynamic and metabolic pathways, as well as interactions between vasoactive molecules, growth factors, and cytokines. Accumulating experimental and clinical evidence suggests that podocytes are highly vulnerable to oxidative damage, and increased oxidative stress appears to be the last and common pathway shared by various aggressors at the cellular level.
A key event in the development and progression of focal segmental glomerulosclerosis (FSGS) is the activation of transforming growth factor beta – a protein (representative of cytokines) TGF- β in podocytes. Recent evidence suggests that activation of transforming growth factor beta TGF- β is associated with enhanced cross-interaction between podocytes and glomerular endothelium through endothelin signaling. The result of the interaction of endothelin with its receptors is the suppression of mitochondrial function and the induction of oxidative stress in the endothelium of the glomeruli, while oxidative damage to mitochondrial DNA becomes evident before podocyte damage. Moreover, specific endothelin-1 and its receptor antagonists or antioxidants targeting mitochondria appear to reverse mitochondrial oxidative stress, endothelial cell dysfunction, and podocyte depletion in this experimental model of focal segmental FSGS glomerulosclerosis. The mechanisms by which endothelial mitochondrial damage and cellular dysfunction promote podocyte apoptosis and progression of FSGS glomerulosclerosis to overt renal failure remain to be elucidated; however, this may be due to decreased levels of nitric oxide NO, which occurs in diabetic neuropathy [Petra et al., 2019].
In the glomeruli, reactive oxygen species ROS are known to mediate cell proliferation and cause glomerular hypertrophy and scarring, impairing renal function. Reactive oxygen species ROS are also associated with upregulation of chemokines and cytokines through the activation of the nuclear factor κB NF-κB transcription factor and hence inflammation. Indeed, patients with type-1 cardiorenal syndrome exhibit higher levels of circulating IL-6 and reactive oxygen species ROS compared to patients with acute heart failure [Lv et al., 2018].
These data support the fact that proteinuric kidneys are highly susceptible to oxidative stress. In addition, severe proteinuria is associated with increased glomerular filtration of plasminogen, which is activated to plasmin by the urokinase-type-plasminogen activator (uPA). Human podocytes express plasminogen receptors and urokinase-type plasminogen uPA. It has been shown that treatment of podocytes with plasminogen increases the regulation of nicotine amide adenine dinucleotide phosphate oxide-dependent superoxide NADPH oxidases NOXX2 and NOX4 isoforms and increases the production of free radicals, especially superoxide anion, by mitochondria [Caio-Silva et al., 2020]. Superoxide anion promotes the synthesis of endothelin-1 and the expression of the β-scavenger receptor CD36, which leads to apoptosis of podocytes.
2.5. Fibrosis
Kidney fibrosis is the last common pathological denominator in chronic kidney damage that occurs regardless of the underlying disorder. The degree of tubulointerstitial fibrosis is the best indicator of renal survival in patients with CKD. Kidney fibrosis is characterized by excessive deposition of extracellular matrix, which destroys and replaces the normal renal parenchyma and, consequently, leading to progressive loss of renal function. The renal scarring process, more understood in recent years, involves a complex interaction of molecular pathways, growth factors, cytokines, and cells [Djudjaj, Boor, 2019].
Kidney myofibroblasts, derived from resident renal fibroblasts and hematopoietic cells migrating to the kidney, are key collagen-producing cells that are involved in fibrosis. Transforming growth factor beta 1 TGF- β1 is a key molecule responsible for myofibroblast differentiation into a profibrotic phenotype characterized by α-smooth muscle actin (αSMA) expression and contractile properties. Both isoforms of nicotine amide adenine dinucleotide phosphate oxide-dependent superoxide NADPH oxidase and their byproducts such as superoxide appear to play a dominant role in the phenotypic transition of fibroblasts to myofibroblasts and fibrogenesis [Chu et al., 2017].
Nox4 oxidase has been recognized as an important mediator in uremic toxin-induced epithelial damage in the proximal renal tubules. More specifically, experimental models show that a p-cresyl sulfate, a uremic toxin that accumulates with the progression of CKD, increases the activity of No4-, p22phox-NADPH oxidase and the production of reactive oxygen species ROS in renal tubular cells, which in turn induces the expression of inflammatory cytokines and profibrotic factors, which ultimately lead to a decrease in cell viability [Chu et al., 2017; Li et al., 2017; Gonzalez-Vicente et al., 2019]. Likewise, indoxyl sulfate, another uremic toxin that accumulates as early as stages 3-4 of CKD, is associated not only with the progression of glomerulosclerosis and interstitial renal fibrosis, but also with cardiac fibrosis.
Autophagy is a physiological process that involves the degradation and recycling of intracellular components and serves as an auxiliary mechanism for the removal of damaged or old macromolecules or organelles. It is believed that autophagy plays a protective role in both acute and chronic kidney disease. New data suggest that oxidative stress autophagy are interrelated, and that reactive oxygen species ROS together with reactive nitrogen species (RNS) induce autophagy and vice versa [Caio-Silva et al., 2020].
Thus, it is assumed that fibrosis is one of the main factors of cardiorenal syndrome, but treatment aimed at this pathophysiology remains clinically unmet. In general, the regulation and dysregulation of collagen synthesis and degradation (and therefore of the extracellular matrix) are fundamental to alleviate physiological and pathological fibrosis [Junho et al., 2017]. Collagen synthesis is stimulated by pathophysiology and tissue damage, while collagen degradation is caused by matrix metallopeptidases (TIMPs) [Wong et al., 2019]. An imbalance in the activity of the matrix metallopeptidases and their tissue inhibitors leads to pathological fibrosis. These events are also associated with the trans-differentiation of vascular smooth muscle and endothelial cells into a profibrotic phenotype (i.e. myofibroblasts), which is also well characterized in cardiac and renal fibroblasts. Fibrosis is also mediated by a variety of intracellular and extracellular growth factors and cytokines.
Transforming growth factor beta 1 TGF- β1 is one of the most prominent growth factors involved in the production, proliferation and differentiation of the extracellular matrix, as well as in immune modulation, Angiotensin 2 Ang II has profibrotic effects through the activation of transforming growth factor beta 1 TGF- β1, and the Ang II-TGF- β1 interaction is associated with chronic hypertension and myocardial fibrosis. Aldosterone is associated with cardiorenal matrix metallopeptidases and tissue inhibitors of metalloproteinase imbalance through activation of nuclear factor κB NF-κB and transforming growth factor beta1 TGF- β1. In addition, uremic toxins, chronic inflammation, and metabolic disorders such as dyslipidemia are also associated with cardiac, renal, and vascular fibrosis by similar mechanisms.
2.6. Dysfunction of the endothelial vascular system
The endothelium is an important component in the regulation and maintenance of normal kidney function. It is well known that the vascular endothelium is particularly vulnerable to oxidative stress, and vascular oxidative stress is thought to play a critical role in the progression of CKD. One of the most important functions of the endothelium is the release of nitric oxide (NO), a relatively unstable diatomic free radical involved in several biological processes, including vasodilation mediated by cyclic guanosine monophosphate (cGMP) in smooth muscle cells, inflammation and immune responses [Fujii et al., 2019]. Nitric oxide NO is synthesized from arginine by the enzyme nitric oxide synthase (NOS), which is expressed in various isoforms-inducible (iNOS), constitutive (cNOS), neural (nNOS), and endothelial (eNOS) – in endothelial cells and some other cell types. Accordingly, in renal tissue, the constitutive isoform (cNOS) is expressed in vessels, glomeruli, and tubules; isoform inducible iNOS is mainly found in vascular smooth muscle cells (VSMC) and mesangium; whereas the endothelial isoform of eNOS is specifically associated with the vascular endothelium. In endothelial cells, eNOS is present on the cell membrane and on the cytosolic surface of the outer mitochondrial membrane. Nitric oxide NO is considered a key molecule directly involved in the pathogenesis of renal failure mediated by oxidative stress [Liu H.J., Liu B., 2018]. The relationship between nitric oxide NO and reactive oxygen species ROS is bidirectional, with low levels of nitric oxide NO in the endothelium triggering the expression of antioxidant genes and protecting renal endothelial and mesangial cells from apoptosis and fibrosis, while, on the other hand, increased levels of reactive oxygen species ROS decreases endothelial production [Sárközy et al., 2018; Medvedeva, 2021]. Under normal conditions, nitric oxide NO in cells is assumed to inhibit cytochrome C oxidase, a mitochondrial membrane-bound terminal enzyme in the electron transport chain, thereby potentially altering the production of mitochondrial reactive oxygen species by ROS. Subsequently, renal endothelial dysfunction and increased vascular resistance occur with a loss of the ability of nitric oxide NO to induce vasodilation and to balance vasoconstrictors such ass angiotensin II, endothelin-1, and sympathetic nervous system factors. In addition, peroxynitrites cause further tissue damage by interacting with various target molecules, including thiols, lipids, and proteins containing aromatic amino acids.
Thus, oxidative stress and inflammation, as well as their interaction, are considered the main pillars of the pathogenesis and progression of CKD (Fig. 2) [Dhaun et al., 2006]. Oxidative stress promotes inflammation through the formation of proinflammatory oxidized lipids, AOPP and AGE (advanced glycation end products), while activation of the nuclear factor κB (NFκB) transcription factor in the prooxidant environment promotes the expression of proinflammatory cytokines as well as the activation of leukocytes and other resident proinflammatory cells. Similarly, pro-inflammatory cytokines such as tumor necrosis factor- α (TNFα) bind to their receptors on tubules and other renal cells and trigger signaling pathways that activate nuclear factor κB (NFκB) transcription factors. In addition, in conditions of chronic inflammation, activated leukocytes generate reactive oxygen species ROS, chlorine and nitrogen, thus enhancing and maintaining oxidative stress. Indeed, initial renal damage mediated by ROS oxidative stress triggers the subsequent renal and systemic inflammatory response.
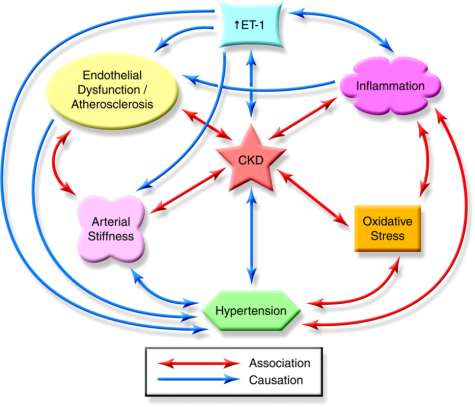
Fig. 2. The role of endothelin-1 in chronic kidney disease (CKD) and cardiovascular disease. ET-1 – endothelin-1 [Dhaun et al., 2006]
Рис. 2. Роль эндотелина-1 при хронической болезни почек (ХБП) и сердечно-сосудистых заболеваниях. ЭТ-1 – эндотелин-1 [Dhaun et al., 2006]
2.7. Role of uremic toxins
Intestinal dysbiosis is observed both in patients with heart failure and in patients with chronic kidney disease. Intestinal dysbiosis caused by chronic kidney disease is thought to be the result of altered nutrient bioavailability and an increase in luminal pH due to increased ammonia levels. It is unclear whether intestinal dysbiosis is a potential “cause” or a consequence of cardiorenal syndrome. Nevertheless, there is no doubt that the gut-heart-kidney axis exists and is of great importance for the treatment of cardiorenal syndrome, which is now being actively studied. One of the key factors affecting intestinal dysbiosis in cardiorenal syndrome is the accumulation of uremic toxins, by-products of the microbial metabolism of food proteins in the intestine. Impaired renal excretory function leads to the accumulation of these solutes, which affect the heart, kidneys and blood vessels [Nallu et al., 2017].
3. Diagnostic and prognostic measures
Traditional cardiac indicators such as leukocytosis, C-reactive protein, and troponins are nonspecific markers of the myocardium [Fan et al., 2018]. For the kidney, serum creatinine and calculated GFR play a central role in the diagnosis of renal injury. Clinical understanding for interpretation and widespread availability, but lack precision in terms of injury site or disease type supports these markers. Cardiac troponin is an excellent marker of myocardial injury and can predict cardiovascular and all-cause mortality in patients with chronic kidney disease and end-stage renal failure (ESRF) [Fu et al., 2018].
IL-18, a soluble tumorigenicity inhibitor 2, a kidney damage molecule [Ronco, Di Lullo, 2014]. Cystatin-C, lipocalin associated with neutrophil gelatinase, sodium uretic peptides are new, promising and highly specialized biomarkers that are sensitive to both kidney and heart disease. Cystatin-C is also predictive of mortality from heart failure and is a good predictor of kidney damage.
More recently, urinary CNP has been identified as a potential biomarker for kidney remodeling and damage in cardiovascular disease [Chen et al., 2019]. MicroRNA (miRNA) is another potentially useful new biomarker in the setting of cardiorenal syndrome with unique profiles specific to heart, kidney and vascular function [Huang et al., 2018; Wang et al., 2018].
Overall, given the complex pathophysiology of cardiorenal syndrome, a multi-marker approach is likely to be required to obtain a better diagnostic/prognostic profile. New biomarkers are limited by their low availability in the clinic and the lack of universal recommendations on cut-off values and proper use.
3.1. Visualization modes
Imaging techniques play an important role in cardiorenal syndrome to assess the structure and function of organs for diagnostic purposes, as well as in research. Ultrasound examination of the kidneys and heart is most often used in practice [Efremova et al., 2019].
MRI is useful for assessing ventricular size, function, and fibrosis, renal blood flow, and renal oxygenation [Chacon-Portillo et al., 2021].
Renal CT, coronary angiography of the heart, PET are rarely used to assess renal function due to contrast-induced renal toxicity [Grande et al., 2017; Hur et al., 2017].
3.2. External and implanted devices
Bioelectric devices are just beginning to enter healthcare practice. Used as an indicator of rehydration in hemodialysis patients. Intra-abdominal pressure measurements can be obtained using a bladder catheter equipped with a transducer. However, such devices are yet to be evaluated in the case of cardiorenal syndrome [Vamos et al., 2018; Rangaswami et al., 2019].
4. Treatment methods
Of course, treatment of the underlying disease is key to cardiorenal syndrome. In case of kidney inflammation, this includes early antibiotics, antispasmodics, detoxification therapy, if necessary, hormones, and vasopressor therapy. Attention should also be paid to anemic conditions in patients with cardiorenal syndrome [McCullough, 2021]. Although in studies, drugs that stimulate erythropoiesis did not improve cardiovascular outcomes in a cohort of patients with chronic kidney disease. However, iron supplementation is encouraged and has been shown to improve symptoms and hospitalization in patients with heart failure [Orvalho, Cowgill, 2017].
Decongestants are needed to regulate volume overload and increased intra-abdominal pressure, and to relieve symptoms. Diuretics remain the primary treatment for volume overload, especially in cardiorenal syndrome of cardiac origin [Rubinstein, Sanford, 2019]. Patients with cardiorenal syndrome may require a much higher dose due to diuretic resistance. High doses of diuretics, of course, leads to increased urine output and a decrease in edema, but can cause temporary impairment of renal function. Inotropes are useful for treating hypotension and low cardiac output [Cowger, Radjef, 2018]. For patients with congestive symptoms without a significant decrease in cardiac output, inotropes are potentially proarrhythmic, have no survival benefit, and should be avoided. Dobutamine and milrinone improve cardiac index in proportion to renal blood flow, although their effect on mortality or clinical outcome is unclear. The combination of low-dose dopamine and a diuretic, as well as levosimendan (a PDE inhibitor), has shown conflicting results in various studies in terms of functional improvement of the kidneys. β-adrenergic receptor blockers may be helpful in improving renal blood flow. Adjunctive use of beta-blockers (metoprolol, bisoprolol, and nebivolol in heart failure and carvedilol in patients with end-stage renal failure) is renoprotective and is associated with reduced hospital admissions and mortality [Rangaswami et al., 2019]. It should be noted that β-blockers are contraindicated in decompensated heart failure due to the potential side effects of hypotension and bradycardia. However, their withdrawal is associated with mortality and therapy should be continued if considered possible. However, their withdrawal is associated with mortality and treatment should be continued if considered possible.
Inhibition of the renin-angiotensin system is one of the main pharmacological treatments for CRS. Angiotensin II receptor antagonists (AT 2) and angiotensin-converting enzyme (ACE-Is) inhibitors are known to cause temporary and reversible impairment or renal function; therefore patient monitoring is necessary [Rubinstein, Sanford, 2019]. The use of these drugs is recommended with the lowest dose, while avoiding non-steroidal anti-inflammatory drugs. RAAS inhibitors can also be limited to hyperkalemia, which can lead to harmful arrhythmias [Clegg et al., 2017]. Given the therapeutic success of sacubitril-valsartan, which increases the natriuretic peptide, natriuretic peptide analogs such as cenderitide, a drug undergoing preliminary preclinical and clinical development, have now been developed [Jing et al., 2017]. In addition, inhibitors of the sodium glucose cotransporter type 2, increasing the excretion of glucose by the kidneys, have recently been of great clinical interest for the treatment of cardiorenal syndrome.
In addition to inhibiting the RAAS, other forms of treatment are needed that can be used alongside the standard regimen. Savira F., et al. Suggest that pharmacological inhibition of ASK1 kinase may help treat cardiorenal syndrome [Savira et al., 2020].
Mitigation strategies for uremic toxins PBUT can be summarized as follows: inhibition of PBUT production in the intestine; inhibition of targeting pathways activated by PBUT that have entered cells; and improved dialysis techniques to remove PBUT [Savira et al., 2020].
A low protein diet has been shown to limit internal production of uremic solutes, which lowers indoxyl sulfate levels in patients with chronic kidney disease [Black et al., 2018]. However, there are also health problems with prolonged low protein intake. The use of symbiotic therapy (prebiotic and probiotic) showed a positive modification of the stool microbiome and a decrease in the level of circulating indoxyl sulfate; but further evaluation is required in a larger study. AST-120, a carbon adsorbent of indole (a precursor of indoxyl sulfate), has shown tremendous potential as a therapeutic strategy agai8nst protein-bound uremic toxins [Cha et al., 2016]. However, its application requires further research.
Lipid metabolism is an integral part of cardiac function. Oxidized LDL levels are markedly elevated in patients with chronic kidney disease, probably due to increased oxidative stress [Peterson et al., 2020].
The main lipid-lowering treatment is the administration of statins (HMG-CoA reductase inhibitors). HDL agonists such as niacin and fibrates are also promising lipid-lowering therapy strategies. Fibrates are commonly prescribed to modulate triglycerides, but there are far fewer clinical studies involving fibrates than statins. Fibrates are beneficial in reducing the risk of cardiovascular disease in mild to moderate chronic kidney disease, but the effect is unclear in the later stages. Niacin is not excreted by the kidneys and may be safe for chronic kidney disease, but is poorly tolerated. PCSK9 (monoclonal antibody) inhibitors have been shown to be more effective than statins in reducing LDL cholesterol and cardiovascular events [Dincer et al., 2019].
An imbalance of sphingolipids can have negative consequences for the heart, kidneys and blood vessels. It has been suggested that RAAS, especially angiotensin II interacts with sphingolipids, although the mechanisms remain unclear. Further research detailing the behavior of the various forms of sphingolipids is needed to understand the benefits of sphingolipid-targeted therapy for diseases including cardiorenal syndrome [Magaye et al., 2019].
Thus, the pathophysiology of CRS is complex and includes interactions between neurohormones, inflammatory processes, oxidative stress, and metabolic disturbances. Therapy remains inadequate currently, mainly symptomatic therapy with minimal chances of full recovery.
Conclusion
Cardiorenal syndrome, which has a complex and multifactorial pathophysiology, is a clinical problem. Diagnostic, prognostic and therapeutic measures for cardiorenal syndrome are limited. Current pharmacological treatments are effective but insufficient to satisfactorily treat or mitigate the progression of cardiorenal syndrome, so they are a high priority area for drug discovery and new therapeutic strategies. Treatment of patients with cardiorenal syndrome should be comprehensive and continuous, aimed at eliminating physical and psychosocial symptoms.


Список литературы
Список использованной литературы появится позже.